FeatureCounts 是一款快速且准确的将测序 reads 分配到基因组特征(如基因、外显子等)的软件,主要用于 RNA-Seq 数据的定量分析。它能够基于与参考基因组的比对结果,统计每个基因或转录本上的测序 reads 数,从而反映基因的表达水平。 功能特点 高效快速:FeatureCounts 采用了优化的算法,能够在短时间内处理大规模的测序数...
featureCounts是一个通用的read定量工具,它将映射reads (RNA-seq reads或基因组DNA-seq reads) 分配给基因组features或meta-features。features是一个参考序列上的区间 (位置范围)。meta-features是表示感兴趣的生物结构的一组features。例如,features通常对应于外显子,meta-features对应于基因。在GTF或SAF注释中共享相同...
featuresCounts软件用于统计基因/转录本上mapping的reads数,也就是用于raw count定量。该软件不仅支持基因/转录本的定量,也支持exon, gene bodies, genomic bins, chromsomal locations等区间的定量。 官网如下 http://bioinf.wehi.edu.au/featureCounts/ featureCounts集成在subreads 软件中, 类似 word 和 office 的关...
featureCounts,有两个核心概念: Feature: 指的是基因组区间的最小单位,比如exon; Metafeature: 可以看做是许多的feature构成的区间,比如属于同一个gene的外显子的组合。 在定量的时候,支持对单个feature 定量(对外显子定量), 也支持对meta-feature进行定量(对基因进行定量)。当reads比对到2个或者以上的features 时...
FeatureCounts的定量原理如下: 1.特征注释:FeatureCounts首先需要使用注释文件来确定基因的位置。注释文件可以是GTF、GFF或SAF格式的文件,其中包含基因的位置和其他相关信息。 2.读取比对:接下来,FeatureCounts需要输入RNA-Seq数据的比对结果文件。常见的比对工具有TopHat、STAR和HISAT等。比对结果文件通常为SAM或BAM格式。
Feature指的是基因组中的最小单位,如外显子,而Metafeature则由多个Feature构成,如属于同一个基因的外显子集合。在定量时,支持对外显子单个Feature进行量化,也支持对Metafeature进行量化,即对整个基因进行定量。当reads匹配到两个或更多的Features时,featureCounts默认忽略这部分reads。若希望统计这部分...
从下图中可看出,两者的数据是呈正相关,两者绝大部分的counts数是非常接近的,在低表达量的那部分数据中HTSeq-count的值偏小于featureCounts。因而至少这个数据来说,我觉得featureCounts代替HTSeq-count进行表达量定量是完全没问题的。 featureCount_htseq-count ...
featureCounts允许在特征或meta-feature级别进行read总结。建议使用独特的基因标识符(如NCBI Entrez基因标识符)将特征聚类为meta-feature。在RNA-seq实验中,通常不计算与一个以上基因重叠的read或片段,因为任何单个片段都必须仅来源于一个靶基因,但无法确定真正靶基因的身份。然而,在ChIP-seq实验中,多重...
转录组定量-featureCounts 不同参数的结果比较: 1 featureCounts -p -Q 10 -s 0 -T $cpu -a $gtf-o $out_dir/${sName}.all.counts.txt $out_dir/${sName}.hg19Aligned.out.bam 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 || Threads ...
基因/转录本/任意特征 表达定量工具之featureCounts使用方法 | 参数详解 2018-03-27 17:49 −... Life·Intelligence 0 13876 ArrayList实现原理(JDK1.8) 2019-11-30 19:14 −### ArrayList实现原理(JDK1.8) 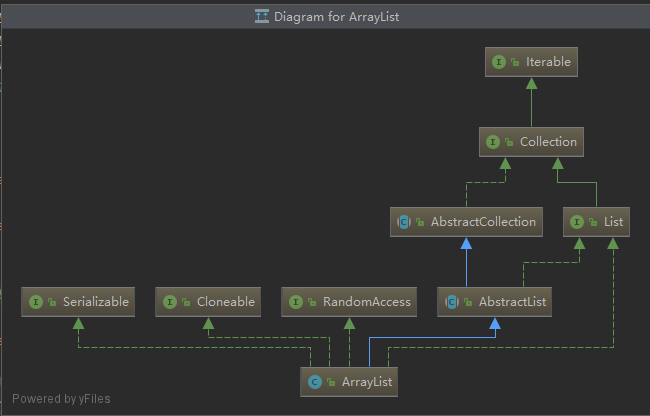...