下面是一些常见的featurecounts参数及其含义: 1. -t <feature_type>:设置要计数的特征类型,如gene、transcript、exon等。默认为gene。 2. -g <attribute_type>:设置要使用的特征属性类型。对于基因,可以使用gene_id、gene_name等;对于转录本,可以使用transcript_id、transcript_name等。默认为gene_id。 3. -a <...
分析参数指的是FeatureCounts进行基因表达分析时所使用的统计方法和计数方法等。在FeatureCounts中,分析参数有以下几个: -s或--strandness。指定测序方向。默认为0(自动检测),1(反义链),2(正义链)。 -m或--minOverlap。指定reads与特征的最小重叠长度。默认为1(至少有1个碱基重叠)。 -M或--maxOverlap。指定read...
转录组定量-featureCounts 不同参数的结果比较: 1 featureCounts -p -Q 10 -s 0 -T $cpu -a $gtf-o $out_dir/${sName}.all.counts.txt $out_dir/${sName}.hg19Aligned.out.bam 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 || Threads ...
所以如果你在你的featureCounts中添加了-p但是没有添加--countReadPairs参数,得到的结果依然会是按照单端测序进行reads计数 #正确的方法是加上--countReadPairs featureCounts -T 5 -p --countReadPairs -t exon -g gene_id \ -a $grch38_annotation_gtf -o all.id.txt *.bam 1>counts.id.log 2>&1 &...
基因/转录本/任意特征 表达定量工具之featureCounts使用方法 | 参数详解 2018-03-27 17:49 −... Life·Intelligence 0 13876 ArrayList实现原理(JDK1.8) 2019-11-30 19:14 −### ArrayList实现原理(JDK1.8) 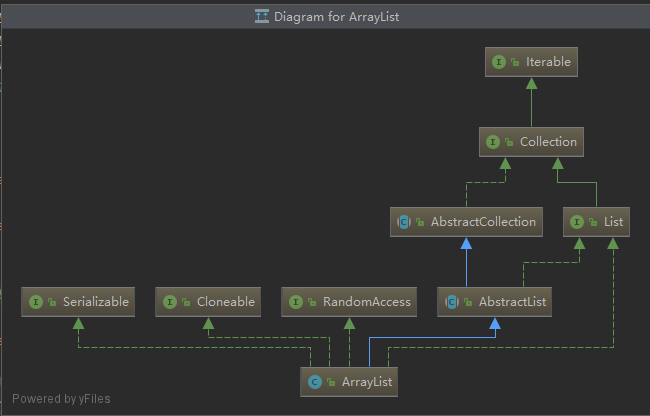...
转录组定量-featureCounts 不同参数的结果比较: 1 featureCounts -p -Q 10 -s 0 -T $cpu -a $gtf-o $out_dir/${sName}.all.counts.txt $out_dir/${sName}.hg19Aligned.out.bam 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 || Threads ...