出现OK表示对应的FASTQ完整。 小结 FASTQ文件由4行为一组构成,第二行和第四行分别为序列信息及序列质量,序列质量可以根据ASCII值表进行查询,传输前后一定要做好md5检验。
如果处理久远一点的FastQ文件需要注意不同测序平台和版本Phred值存在差异,现在基本都是Phred+33体系。 2.vcf文件格式 vcf文件是怎么得到的,一般应用在什么地方: 变异检测工具(如GATK)的输出文件;变异注释(如SnpEff)的输入文件;VCFtools处理工具的处理对象。 文件内容包括注释部分(#开头)和主体部分(site records),各列...
以Illumina测序仪为例,文件通常以样本名、序列号、lane编号、read编号和扩展名结尾,例如:Samplexx_SXX_L002_R1_001.fastq.gz。具体命名包含样本名、样本顺序、lane编号与read编号。"Undetermined_S0_L001_R1_001.fastq.gz"则代表未匹配index的reads。FASTQ文件解读:文件结构以四行为一组,具体信息如...
fasta与fastq格式文件解读 1、FASTA文件的格式 在生物信息学中,FASTA格式(又称为Pearson格式)是一种基于文本的、用于表示核苷酸序列或氨基酸序列的格式。在这种格式中碱基对或氨基酸用单个字母来表示,且允许在序列前添加序列名及注释。 FASTA文件以序列表示和序列作为一个基本单元,各行记录信息如下: 第一行是由大于号"...
首先,FastQ格式用于存储测序读取数据,每条reads信息由四行组成,包括序列标识、序列本身、质量值标识,以及详细的位置和测序信息。其质量值通过ASCII值代表测序错误概率,Phred+33或Phred+64体系中的较高质量值(如Q20和Q30)表明较低的错误率。双端测序的FastQ文件,其header行可能显示序列来源的区别。vcf...
fastq文件格式解读 二代测序返回的结果有时候一个物种的测序结果会返回来两个双端的fastp。 r1.fq.gz l1.fq.gz r2.fq.gz l2.fq.gz 测序数据内容实际上一块的,只是传输时分成两个部分。 我们使用时习惯将其合并为一个双端文件。 原理 原理就是将两个文件内容依次输入到一个新的文件内,你也可以将第二个...
原博文 fasta与fastq格式文件解读 2017-05-08 14:30 −... lulu.bioinfo 0 4534 ArrayList实现原理(JDK1.8) 2019-11-30 19:14 −### ArrayList实现原理(JDK1.8) 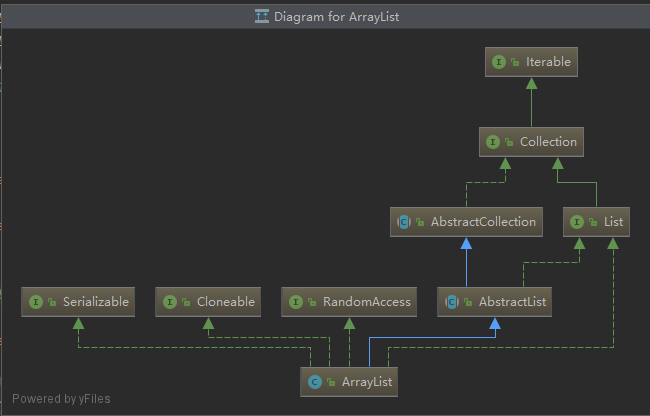 ``` java public class ArrayLis... ...